Research Report
A New Perspective on Revealing Tumor Heterogeneity through Single Cell RNA Sequencing 
 Author
Author  Correspondence author
Correspondence author
Cancer Genetics and Epigenetics, 2024, Vol. 12, No. 1 doi: 10.5376/cge.2024.12.0007
Received: 29 Dec., 2023 Accepted: 02 Feb., 2024 Published: 15 Feb., 2024
Chen T., 2024, Single-cell RNA sequencing reveals new insights into tumor heterogeneity, Cancer Genetics and Epigenetics, 12(1): 55-65 (doi: 10.5376/cge.2024.12.0007)
This review paper mainly explores the application and prospects of single-cell RNA sequencing technology in the study of tumor heterogeneity. As an important concept in the field of oncology, tumor heterogeneity reveals the diversity and complexity of cells within a tumor, which has significant implications for tumor initiation, progression, treatment, and prognosis. However, traditional research methods have limitations in elucidating tumor heterogeneity. In recent years, the emergence of single-cell RNA sequencing technology has brought new breakthroughs to the study of tumor heterogeneity. This paper first introduces the concept and importance of tumor heterogeneity, then outlines the development and principles of single-cell RNA sequencing technology. Subsequently, it focuses on the specific applications of single-cell RNA sequencing in tumor heterogeneity research, including the identification of intra-tumoral cell subpopulations, the analysis of gene expression differences and regulatory networks, and the investigation of interactions among tumor cells. Finally, the contributions of single-cell RNA sequencing in tumor heterogeneity research are summarized, and future research directions are prospected.
Tumor heterogeneity is a core and complex issue in contemporary oncology research. It refers to the significant differences in genetics, epigenetics, metabolism, and cell behavior between different cell populations within the same tumor. This heterogeneity not only affects the growth rate, invasion ability and response to treatment of tumors, but also greatly increases the risk of disease recurrence and metastasis, making precision treatment of tumors facing huge challenges (Stanta and Bonin, 2018). Therefore, a deep understanding of the nature and sources of tumor heterogeneity is crucial for the development of more effective diagnostic and therapeutic strategies.
In recent years, with the rapid development of biotechnology, single-cell RNA sequencing (scRNA-seq) technology has emerged and gradually matured. This technology allows researchers to make high-precision measurements of genome-wide transcription at the level of individual cells, revealing heterogeneity within cell populations with unprecedented resolution. Compared with traditional cell population-based sequencing methods, scRNA-seq avoids the interference of population average effects and can more accurately depict the unique status of each cell and its dynamic changes throughout the biological process (Papalexi and Satija, 2018).
In the field of oncology, the introduction of scRNA-seq technology provides new opportunities for comprehensive analysis of tumor heterogeneity. By measuring the gene expression profile of individual tumor cells, researchers can not only identify different cell subpopulations within the tumor, but also further analyze the gene expression differences, signaling pathway activation status, and potential intercellular communication between these subpopulations. Mechanism (Ferrall -Fairbank and Meghan, 2019). This information is of great significance for revealing the mechanisms of tumorigenesis, key regulatory networks during development, and mechanisms of response and resistance to treatment strategies.
This study aims to use single-cell RNA sequencing technology to deeply explore the molecular basis of tumor heterogeneity, in order to provide new ideas and strategies for precise diagnosis and treatment of tumors. Through this research, we hope to gain a more comprehensive understanding of the formation and maintenance mechanisms of tumor heterogeneity, thereby providing a scientific basis for the development of targeted therapies for specific cell subpopulations, and ultimately improving the therapeutic effects and quality of life of tumor patients.
1 Traditional Research Methods and Limitations of Tumor Heterogeneity
1.1 The concept of tumor heterogeneity
Tumor heterogeneity (Figure 1) is an extremely critical and complex issue in the field of contemporary oncology. It refers to the genetics, phenotypic characteristics, metabolic activity, and response to treatment among different cells within a single tumor. significant differences across dimensions. This heterogeneity not only runs through the entire process of tumor occurrence, development, and metastasis, but is also an important factor affecting patient prognosis and treatment response.
.gif) Figure 1 The composition and impact of tumor heterogeneity (Bai et al., 2020) |
.gif){kind=link}
From a genetic perspective, tumor heterogeneity is reflected in differences in gene mutations, copy number variations, and epigenetic modifications between different cells. These genetic variations may confer different growth advantages, invasion capabilities, and drug sensitivity to tumor cells (Wang et al., 2020). In addition, phenotypic heterogeneity is manifested in the diversity of tumor cells in terms of morphology, differentiation status, metabolic activity, and intercellular communication. This phenotypic heterogeneity is driven in large part by genetic heterogeneity as well as the complex interplay of the tumor microenvironment.
The existence of tumor heterogeneity brings great challenges to the diagnosis and treatment of tumors. In his article, Welch (2016) emphasized the pervasiveness of heterogeneity in all cancer types, including genetic intrinsic, epigenetic, positional, and population-level heterogeneity, and explored these properties have profound implications for understanding tumor behavior and how (or should be) treated. First, because different cell subpopulations may respond very differently to treatment, traditional “one-size-fits-all” treatments often fail to completely eliminate all tumor cells, leading to disease recurrence and progression. Secondly, tumor heterogeneity is also one of the important reasons leading to drug resistance. During treatment, drug-sensitive cells may be selectively killed, while drug-resistant cell subpopulations may gradually dominate, leading to treatment failure.
Therefore, a deep understanding of the nature and sources of tumor heterogeneity is crucial for the development of more effective diagnostic and therapeutic strategies. Through the study of tumor heterogeneity, it is expected to reveal the molecular mechanisms of tumor occurrence and development, discover new therapeutic targets, and provide scientific basis for precise tumor treatment.
1.2 Overview of traditional research methods
In the process of deeply exploring tumor heterogeneity, traditional research methods play an important role, although they have certain limitations. These methods mainly rely on macroscopic and microscopic analysis of tumor tissue samples, as well as the application of molecular biology techniques.
Dagogo-Jack and Shaw (2018) discuss the nature of cancer as a dynamic disease, which often becomes more heterogeneous as the disease progresses. Because of this heterogeneity, the entire tumor may contain a diverse collection of cells carrying different molecular signatures that vary in sensitivity to treatment. This heterogeneity may result in non-uniform distribution of genetically distinct tumor cell subpopulations within and outside the disease site (spatial heterogeneity) or temporal changes in the molecular makeup of cancer cells (temporal heterogeneity).
At the histological level, traditional methods include morphological observation under a light microscope, such as hematoxylin-eosin staining (H&E staining) to evaluate the morphology, arrangement, and differentiation of tumor cells. Immunohistochemistry (IHC) further uses specific antibodies to detect protein expression in tumor cells, thereby revealing the existence of different cell subpopulations and their molecular characteristics. These methods can provide important information on the spatial distribution and cellular composition of tumor tissues (Luecken and Theis, 2019).
At the molecular biology level, traditional research methods mainly focus on expression analysis of genes and proteins. For example, gene expression profiling detects the expression levels of thousands of genes in tumor samples using microarray or quantitative PCR techniques to identify gene groups associated with specific cell subpopulations or biological processes. In addition, mutation screening and genome sequencing are also widely used to identify genetic variations in tumor cells that may be important drivers of tumor heterogeneity.
However, these traditional approaches face some common challenges when dealing with tumor heterogeneity. Cassidy et al. (2015) explored how patient-derived tumor xenograft models (PDX) can maintain the molecular heterogeneity of their original samples. Although PDX models can largely reproduce the multigene structure of human tumors, they cannot fully Explaining heterogeneity in the tumor microenvironment.
1.3 Limitations of traditional methods
Although traditional research methods are widely used in oncology, they have shown obvious limitations in exploring tumor heterogeneity. These limitations mainly stem from the limitations of the technology itself and the complexity of tumor heterogeneity.
Jamal-Hanjan et al. (2015) pointed out that although genomic studies have revealed a complex and heterogeneous clonal landscape of different tumor origins and treatment responses, cancer progression, and risk of disease recurrence, the significance of subclonal mutations, especially in driver genes The mutations in, as well as their evolution over time and response to cancer treatments, have yet to be determined.
Traditional methods often require population analysis of large numbers of cells, thus failing to accurately capture the heterogeneity between individual cells. Because tumors are complex tissues composed of many different cell types, these cells can differ significantly in gene expression, metabolic activity, and response to treatment. However, traditional methods often only provide averaged results and fail to reveal these subtle cell-to-cell differences, leading to information loss and misinterpretation.
Traditional methods are difficult to dynamically track changes in tumor cells in time and space (Heinrich et al., 2021). Tumor heterogeneity is a dynamic process, and with tumor development and therapeutic intervention, cell subpopulations may change, with new subpopulations appearing or old subpopulations disappearing. However, traditional methods often only provide static information and fail to reflect these dynamic changes, thus limiting a comprehensive understanding of tumor heterogeneity.
Traditional methods also have difficulties in identifying and isolating different cell subpopulations within tumors. Due to technical limitations, these methods are often unable to accurately identify different cell types within tumors, let alone further isolate and study the biological properties of these cell subpopulations. This severely limits our understanding of the sources and maintenance mechanisms of tumor heterogeneity. Dagogo-Jack and Shaw (2018) discussed how tumor heterogeneity increases with disease progression, and tumors may contain subpopulations of cells with different molecular characteristics and different sensitivities to treatments. They emphasized the accurate assessment of tumors. The importance of heterogeneity in developing effective treatments.
2 Principles and Advantages of Single-cell RNA Sequencing Technology
2.1 Principles of single-cell RNA sequencing technology
Single-cell RNA sequencing technology (Figure 2), as a major breakthrough in the field of bioinformatics in recent years, its principle integrates cutting-edge technologies in multiple fields such as molecular biology, microfluidic technology, and high-throughput sequencing. This technology aims to solve the bottleneck of traditional RNA sequencing methods that cannot be accurate to individual cells, and provides life science researchers with a new perspective to gain insight into the differences and complexities between cells.
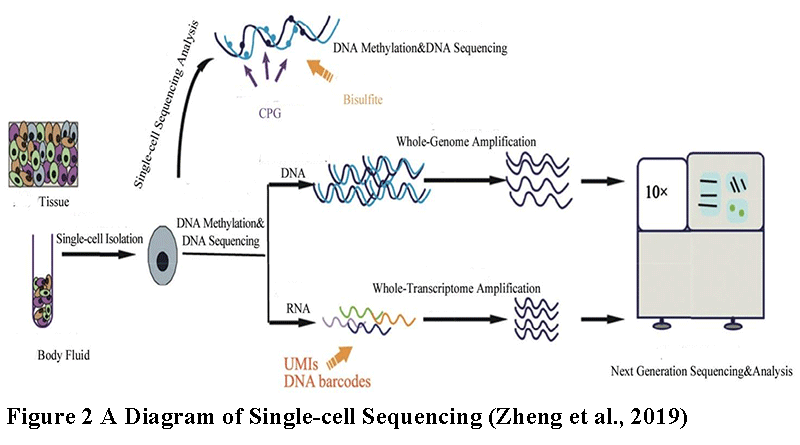 Figure 2 A Diagram of Single-cell Sequencing (Zheng et al., 2019) |
.gif){kind=link}
In the initial stages of single-cell RNA sequencing, single cells need to be efficiently isolated from complex biological samples. This is usually achieved with the help of microfluidic chips or droplet generation systems, ensuring that each processing unit contains only one cell. These isolated individual cells are then lysed, releasing the intracellular RNA molecules.
Next, the released RNA is converted into cDNA by reverse transcriptase. In this step, reverse transcriptase uses RNA as a template to synthesize a complementary DNA strand, called cDNA. This process retains the information of the original RNA but converts it into a more stable form of DNA that is easier to manipulate and sequence later.
Since the RNA content in a single cell is extremely low, it is often difficult to obtain sufficient data by direct sequencing. Therefore, cDNA usually needs to be amplified to increase its quantity before sequencing. This is usually achieved through PCR (polymerase chain reaction) technology, which can exponentially amplify specific DNA fragments in a short time (Picelli, 2017).
Finally, the amplified cDNA is sent to a high-throughput sequencer for sequencing. Sequencers can read the sequence information of millions of DNA fragments simultaneously, thereby generating large amounts of data. These data are then subjected to bioinformatics analysis to reveal key information such as gene expression in individual cells, cell type, functional status, and interaction with other cells.
2.2 Advantages of single-cell RNA sequencing technology
The advantages of single-cell RNA sequencing technology are particularly prominent compared with traditional sequencing methods based on cell populations. In traditional methods, researchers can usually only obtain the average gene expression of a group of cells, and such data often obscures the differences between individual cells. However, every cell in an organism is unique, and small differences between them can be critical to the function and behavior of the entire organism.
The emergence of single-cell RNA sequencing technology has broken this limitation. It can capture the gene expression information of individual cells, thereby revealing the heterogeneity between cells. This heterogeneity plays a key role in complex biological processes, such as embryonic development, immune response, disease development, etc. Through single-cell RNA sequencing, researchers can more accurately understand cell types and status changes during these processes, providing new ideas for the diagnosis and treatment of diseases.
AlJanahi et al. (2018) provide basic principles of the new technology, focusing on important concepts in single-cell RNA sequencing data analysis, such as quality control of data, normalization and standardization methods, and clustering for data dimensionality reduction. and visualization algorithms.
Zhang et al. (2021) study used single-cell RNA sequencing technology to deeply analyze the heterogeneity of cancer cells, revealing the existence of different cell subpopulations and their association with tumor occurrence and development. It provides new ideas and methods for precise diagnosis and treatment of cancer.
Zheng and Wang (2019) studied the application prospects of single-cell RNA sequencing technology in early diagnosis, prognosis assessment and new drug development of tumors, providing an important basis for the formulation of treatment strategies for solid tumors. This research lays the foundation for understanding the complexity of solid tumors and advancing personalized treatments.
Single-cell RNA sequencing technology also has extremely high sensitivity. It is able to detect very rare cell types or low-abundance transcripts that are often difficult to detect with traditional methods. Single-cell RNA sequencing technology is still developing, and combined with other technologies such as spatial transcriptomics and single-cell multi-omics, it provides more powerful tools for life science research. With the continuous advancement of technology and reduction of costs, it is believed that single-cell RNA sequencing technology will play an increasingly important role in future biomedical research.
2.3 Technical challenges and solutions
Although single-cell RNA sequencing technology has brought revolutionary progress to biomedical research, it still faces some technical challenges in practical application. These challenges mainly come from the complexity of single-cell operations, the scarcity of RNA, and the difficulty of data analysis.
The capture and lysis of single cells is a delicate and complex task. Because cells are small in size, they are easily disturbed by the external environment. The RNA content in single cells is extremely low, which brings great difficulties to the reverse transcription and amplification processes. Low-abundance RNA can easily lead to low reverse transcription efficiency and amplification bias, thus affecting the accuracy and reliability of sequencing results (Isoda et al., 2017). In order to overcome these challenges, researchers continue to optimize capture platforms such as microfluidic chips and droplet microfluidic technology to improve the capture efficiency and lysis effect of single cells, and explore adding exogenous RNA or using pre-amplification strategies. method to increase the strength and stability of sequencing signals.
Kiselev et al. (2019) discuss the challenges of unsupervised clustering of single-cell RNA sequencing data, which is a challenge in analyzing these data from a computational perspective, and the aspects with the data that make it challenging.
Vallejos et al. (2017) explore the challenges and opportunities of normalizing single-cell transcriptome data, highlighting how using traditional methods risks producing misleading results, as well as providing alternatives and recommendations for single-cell RNA sequencing users.
The massive data generated by single-cell RNA sequencing technology brings huge challenges to data analysis. How to extract meaningful biological information from huge data and accurately interpret the differences and regulatory relationships between cells are important tasks facing researchers.
3 Application of Single-cell RNA Sequencing in Studying Tumor Heterogeneity
3.1 Identification and classification of cell subpopulations within tumors
Tumor is a complex biological system, and the heterogeneity of its internal structure has always been a difficult and hot topic in medical research. Traditional sequencing methods often can only give the average gene expression of the entire tumor tissue, and cannot accurately depict the characteristics of different cell subpopulations within the tumor. However, it is the differences in these cell subpopulations that determine the tumor's growth rate, ability to invade, and response to treatment.
In this context, single-cell RNA sequencing technology emerged as the times require, opening a new door for the study of cell subpopulations within tumors. This technology is able to capture the gene expression information of single cells and can comprehensively and deeply explore the heterogeneity within tumors under unbiased conditions. Through single-cell RNA sequencing, it is not only possible to discover rare cell subpopulations that are masked in traditional sequencing, but also to accurately identify the unique molecular markers of each cell subpopulation, thereby accurately classifying them.
Wang et al. (2021) conducted a comprehensive analysis of consensus molecular subtypes (CMS) of colorectal cancer (CRC) through single-cell RNA sequencing data, revealing the heterogeneity of gene regulatory networks and identifying key regulators of CRC.
Ye et al. (2020) proposed a new learning framework to detect interactive gene groups in scRNA-seq data based on co-expression network analysis and subgraph learning, providing a systematic gene ontology for the detection of interactive gene groups in different cancer subtypes. Enrichment analysis.
Such as pancreatic cancer research: In a study of pancreatic ductal adenocarcinoma (PDAC), researchers used single-cell RNA sequencing technology to identify different cell subpopulations in the tumor, including type 1 ductal cells and type 2 ductal cells. wait. Type 2 duct cells were found to have significantly higher levels of chromosomal copy number variations (CNV) than other cell types, suggesting that this cell subset plays an important role in the progression of PDAC. Type 2 ductal cells are mainly enriched in functional genes related to cancer, such as cell proliferation, migration, and hypoxia.
This classification is not only based on similarities in gene expression, but also takes into account the functional status, differentiation stage, and potential response to treatment of the cells. Therefore, the classification of cell subpopulations obtained through single-cell RNA sequencing technology has more biological significance and clinical application value. It can help us better understand the pathogenesis of tumors, predict patients' treatment response and prognosis, and even guide the development of personalized treatment strategies (Janiszewska, 2020).
3.2 Analysis of gene expression differences and regulatory networks
In the process of exploring tumor heterogeneity, understanding the differences in gene expression between different cell subpopulations and how these differences are regulated is a crucial step. This understanding not only helps to reveal the mechanisms of tumor occurrence and development, but may also provide ideas for new treatment strategies. Single-cell RNA sequencing technology is a powerful tool for analyzing the expression differences and regulatory networks of these genes (Zeisel et al., 2018).
Traditional gene expression analysis methods often can only give the average expression level of a cell population and cannot accurately reflect the differences between individual cells. Single-cell RNA sequencing technology can measure the transcriptome of individual cells, revealing subtle but important gene expression changes among cell subpopulations. These changes may involve specific transcription factors, signaling pathways, or epigenetic modifications, which together form a complex gene regulatory network.
By comparing the gene expression profiles of different cell subpopulations, it is possible to identify which genes are up-regulated or down-regulated in specific subpopulations, and then speculate on the biological processes in which these genes may be involved. In addition, by combining bioinformatics methods and network analysis technology, the interaction network between genes can also be constructed to reveal the regulatory relationship between them. For example, certain transcription factors may regulate processes such as cell proliferation, apoptosis, or invasion by activating or inhibiting the expression of downstream target genes (Lafzi et al., 2018).
He et al. (2020) performed single-cell RNA sequencing (scRNA-seq) on early-stage lung adenocarcinoma patients with EGFR mutations, revealing the heterogeneous tumor and immune cell populations in the tumor microenvironment (TME), providing a basis for understanding cells in the TME. provide unique insights into the complex interactions between.
Huang et al. (2023) proposed a new gene selection method, automatic associated feature learning (AAFL), to automatically identify different gene features of different cell subpopulations (cancer subtypes), which provides a new way to understand cell heterogeneity and complex tumors. Ecosystems provide important insights.
This in-depth analysis of gene expression differences and regulatory networks not only contributes to a more comprehensive understanding of tumor heterogeneity, but may also provide new targets for therapeutic strategies targeting specific cell subpopulations. In the future, with the continuous development and improvement of technology, single-cell RNA sequencing will play an increasingly important role in the field of tumor research and move towards the goal of personalized medicine.
3.3 Research on interactions between tumor cells
In the complex tumor ecosystem, tumor cells do not exist in isolation. They interact closely with surrounding stromal cells, immune cells, and other types of cells, forming an intricate cellular communication network. These interactions have profound effects on tumor growth, invasion, metastasis, and treatment resistance. Therefore, in-depth study of the interactions between tumor cells is of great significance for revealing the pathogenesis of tumors and developing new treatment strategies (Zeisel et al., 2018).
Single-cell RNA sequencing technology provides new perspectives and methods for studying interactions between tumor cells. Single-cell RNA sequencing technology can capture the transcriptome information of single cells and study the interactions between tumor cells and other cell types at the single-cell level.Through single-cell RNA sequencing, multiple cell types in tumor samples can be sequenced and analyzed simultaneously, including tumor cells, immune cells, stromal cells, etc. In this way, gene expression profiles can be compared between different cell types and key genes and signaling pathways related to cell-cell interactions can be identified. In addition, combining bioinformatics methods and network analysis technology, the interaction network between cells can also be constructed to reveal the communication mechanisms and regulatory relationships between different cell types (Zhang et al., 2021).
Cao et al. (2022) used single cell sequencing (scRNA-seq) technology to study the role of tumor heterogeneity in tumor progression, especially how to track gene expression or mutations in heterogeneous cells by measuring the entire transcriptome of single cells. situation, assess the clonal origins of cancer cells, and determine the selective evolution of different subpopulations of cancer cells.
Zheng et al. (2021) performed single-cell RNA sequencing of T cells from more than 300 patients in 21 cancer types, revealing differences in immune cell types related to tumor characteristics, providing a basis for tumor-infiltrating T cells in the tumor microenvironment. Status and abundance provide a systematic comparison.
These studies not only contribute to a deeper understanding of the interactions between tumor cells and the complexity of the tumor ecosystem, but may also provide new ideas for the development of new treatment strategies. For example, by interfering with the interaction between tumor cells and immune cells, the anti-tumor response of the immune system can be enhanced; by regulating the interaction between tumor cells and stromal cells, processes such as tumor invasion and metastasis can be inhibited.
4 Summary and Outlook
4.1 Research summary and main findings
This study deeply explores the application prospects of artificial intelligence in the field of drug design and the ethical considerations it brings, while focusing on the unique value of single-cell RNA sequencing technology in the study of tumor heterogeneity. Through a systematic literature review, in-depth data analysis, and interdisciplinary research methods, the great potential of artificial intelligence technology in drug design is revealed, as well as the ethical challenges that must be faced along with this development.
Alizadeh et al (2015) discussed the extent of tumor heterogeneity, an emerging topic that researchers are only beginning to understand, exploring how genetic and epigenetic heterogeneity influence tumor evolution and clinical progression.
Research has found that artificial intelligence technology can play an important role in all stages of drug design through powerful computing power and advanced algorithm models. From target identification to molecular screening to clinical trial optimization, artificial intelligence can significantly improve research and development efficiency, reduce costs, and is expected to bring revolutionary breakthroughs to new drug development. However, with the rapid development of technology, issues such as how to ensure that the application of artificial intelligence complies with ethical norms, protects patient privacy, and avoids data abuse have become increasingly prominent (Gawad et al., 2016).
In terms of studying tumor heterogeneity, this study further confirms the great value of single-cell RNA sequencing technology. This technology can comprehensively reveal the heterogeneity of tumors at the single-cell level, including differences in gene expression of different cell subpopulations, the complexity of regulatory networks, and the interaction between tumor cells and the microenvironment. These findings not only contribute to a deeper understanding of tumor pathogenesis, but also provide new ideas for the development of therapeutic strategies targeting specific cell subpopulations or microenvironments.
This study not only provides strong theoretical support and practical guidance for the application of artificial intelligence in drug design, but also brings new perspectives and methods to the study of tumor heterogeneity. With the continuous advancement of technology and the deepening of interdisciplinary cooperation, artificial intelligence and single-cell RNA sequencing technology will play an increasingly important role in future medical research.
4.2 Impact and contribution to the study of tumor heterogeneity
Tumor heterogeneity has always been a core problem in cancer research. It refers to the existence of multiple different cell subpopulations within the same tumor. These subpopulations vary in terms of gene expression, proliferation rate, invasion ability, and response to treatment. There are significant differences. The emergence of single-cell RNA sequencing technology has brought revolutionary breakthroughs to the study of tumor heterogeneity (Ma et al., 2020).
This technology can reveal the gene expression profiles of different cell subpopulations within tumors with unprecedented resolution, and can study tumor heterogeneity at the single-cell level. The application of this technology can not only more accurately identify and classify cell subpopulations within tumors, but also enable in-depth analysis of gene expression differences and regulatory networks between these subpopulations. Through these studies, we can gain a deeper understanding of the pathogenesis of tumors, predict patients' treatment response and prognosis, and even guide the development of personalized treatment strategies.
In addition, by simultaneously sequencing and analyzing multiple cell types in tumor samples, single-cell RNA sequencing technology can reveal the communication mechanisms and regulatory relationships between different cell types, further deepening the understanding of the tumor ecosystem. These studies will provide new ideas for developing treatment strategies targeting specific cell subpopulations or tumor microenvironments, and promote the development of precision medicine and personalized treatment of tumors.
Jamal-Hanjani et al (2015) highlighted the potential impact of tumor heterogeneity in treatment response and resistance, cancer progression and risk of disease recurrence, but the significance of subclonal mutations, particularly in driver genes, and their subsequent The evolution over time, as well as the response to cancer treatment, remains to be determined.
Single-cell RNA sequencing technology has had a profound impact on the study of tumor heterogeneity, providing a powerful tool to reveal the pathogenesis of tumors, develop new treatment strategies, and improve treatment effects.
4.3 Prospects and suggestions for future research directions
Looking to the future, with the continuous advancement of science and technology and the deepening of medical research, artificial intelligence and single-cell RNA sequencing technology will present broader development prospects in the fields of drug design and tumor heterogeneity research.
McGranahan and Swanton (2017) review data and techniques on intra-tumor heterogeneity across different cancer types, as well as the intrinsic dynamics of tumor evolution, constraints, and contingency, highlighting macroevolutionary transitions, often involving large-scale chromosomal changes, Importance in driving tumor evolution and metastasis.
Dagogo-Jack and Shaw (2018) discussed how tumor heterogeneity increases with disease progression, and tumors may contain subpopulations of cells with different molecular characteristics and different sensitivities to treatments. They emphasized the accurate assessment of tumor heterogeneity. The importance of sex in developing effective treatments.
Single-cell RNA sequencing technology is a powerful tool for studying tumor heterogeneity, but its resolution and accuracy still have room for improvement. Future research should be devoted to improving sequencing technology, reducing experimental costs, and improving data analysis capabilities so that it can be more widely used in the analysis of clinical samples. In addition, combining other omics technologies, such as single-cell proteomics and metabolomics, will provide more comprehensive information on tumor heterogeneity (Peng et al., 2019).
Interdisciplinary collaboration is key to advancing both fields. It is recommended to strengthen exchanges and cooperation between computer science, bioinformatics, pharmacy, clinical medicine and other disciplines to jointly promote the application of artificial intelligence and single-cell sequencing technology in the medical field. At the same time, ethics and privacy issues also require interdisciplinary teams to jointly discuss and formulate reasonable laws, regulations and ethical guidelines to ensure that while science and technology progress, basic human rights are protected.Artificial intelligence and single-cell RNA sequencing technology bring unlimited possibilities for drug design and tumor heterogeneity research. It is expected that these technologies will play a greater role in the medical field and make greater contributions to human health and well-being in the future.
Alizadeh A., Aranda V., Bardelli A., Blanpain C., Bock C., Borowski C., Caldas C., Califano A., Doherty M., Elsner M., Esteller M., Fitzgerald R., Korbel J., Lichter P., Mason C., Navin N., Pe'er D., Polyak K., Roberts C., Siu L ., Snyder A., Stower H., Swanton C., Verhaak R., Zenklusen J., Zuber J., and Zucman-Rossi J., 2015, Toward understanding and exploiting tumor heterogeneity, Nature Medicine, 21: 846-853.
https://doi.org/10.1038/nm.3915
PMid:26248267 PMCid:PMC4785013
AlJanahi A., Danielsen M., and Dunbar C., 2018, An Introduction to the Analysis of Single-Cell RNA-Sequencing Data. Molecular Therapy, Methods & Clinical Development, 10: 189 - 196.
https://doi.org/10.1016/j.omtm.2018.07.003
PMid:30094294 PMCid:PMC6072887
Bai R.L., and Cui J.W., 2020, Tumor heterogeneity: the great challenge in precision clinical diagnosis and treatment, Chinese Journal of Clinical Oncology, 47(21): 1081-1087.
Cassidy J., Caldas C., and Bruna A., 2015, Maintaining Tumor Heterogeneity in Patient-Derived Tumor Xenografts, Cancer research, 75(15): 2963-8 .
https://doi.org/10.1158/0008-5472.CAN-15-0727
PMid:26180079 PMCid:PMC4539570
Dagogo-Jack I., and Shaw A., 2018, Tumour heterogeneity and resistance to cancer therapies, Nature Reviews Clinical Oncology, 15: 81-94.
https://doi.org/10.1038/nrclinonc.2017.166
PMid:29115304
Ferrall-Fairbanks M.C., Ball M., Padron E., and Altrock P.M., 2019, Leveraging Single-Cell RNA Sequencing Experiments to Model Intratumor Heterogeneity, JCO Clin Cancer Inform, 3: 1-10.
https://doi.org/10.1200/CCI.18.00074
PMid:30995123 PMCid:PMC6873939
Gawad C., Koh W., and Quake S.R., 2016, Single-cell genome sequencing: current state of the science, Nat Rev Genet, 17(3): 175-188.
https://doi.org/10.1038/nrg.2015.16
PMid:26806412
Heinrich S., Craig A.J., Ma L., Heinrich B., Greten T.F., and Wang X.W., 2021, Understanding tumor cell heterogeneity and its implication for immunotherapy in liver cancer using single-cell analysis, Journal of Hepatology, 74(3): 700-715.
https://doi.org/10.1016/j.jhep.2020.11.036
PMid:33271159
Huang M., Long C., Ma J., 2023, AAFL: automatic association feature learning for gene signature identification of cancer subtypes in single-cell RNA-seq data, Brief Funct Genomics, 22(5): 420-427.
https://doi.org/10.1093/bfgp/elac047
PMid:37122141
He D., Wang D., Lu P., Yang N., Xue Z., Zhu X., Zhang P., and Fan G., 2020, Single-cell RNA sequencing reveals heterogeneous tumor and immune cell populations in early-stage lung adenocarcinomas harboring EGFR mutations, Oncogene, 40: 355 - 368.
https://doi.org/10.1038/s41388-020-01528-0
PMid:33144684 PMCid:PMC7808940
Isoda T., Moore A.J., and He Z., 2017, Non-coding transcription instructs chromatin folding and compartmentalization to dictate enhancer-promoter communication and T cell fate, Cell, 171(1):103-119.
https://doi.org/10.1016/j.cell.2017.09.001
PMid:28938112 PMCid:PMC5621651
Janiszewska M., 2020, The microcosmos of intratumor heterogeneity: the space-time of cancer evolution. Oncogene, 39(10): 2031-2039.
https://doi.org/10.1038/s41388-019-1127-5
PMid:31784650 PMCid:PMC7374939
Jacquemin V., Antoine M., Dom G., Detours V., Maenhaut C., and Dumont J.E., 2022, Dynamic cancer cell heterogeneity: diagnostic and therapeutic implications, Cancers, 14(2): 280.
https://doi.org/10.3390/cancers14020280
PMid:35053446 PMCid:PMC8773841
Jamal-Hanjani M., Quezada S., Larkin J., and Swanton C., 2015, Translational Implications of Tumor Heterogeneity, Clinical Cancer Research, 21: 1258-1266.
https://doi.org/10.1158/1078-0432.CCR-14-1429
PMid:25770293 PMCid:PMC4374162
Kiselev V., Andrews T., and Hemberg M., 2019, Challenges in unsupervised clustering of single-cell RNA-seq data, Nature Reviews Genetics, 20: 273-282.
https://doi.org/10.1038/s41576-018-0088-9
PMid:30617341
Luecken M.D., and Theis F.J., 2019, Current best practices in single‐cell RNA‐seq analysis: a tutorial, Molecular systems biology, 15(6): e8746.
Lafzi A., Moutinho C., and Picelli S., 2018, Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies, Nat Protoc, 13(12): 2742-2757.
https://doi.org/10.1038/s41596-018-0073-y
PMid:30446749
Ma X., Guo J., Liu K., Chen L., Liu D., Dong S., and Zou C., 2020, Identification of a distinct luminal subgroup diagnosing and stratifying early stage prostate cancer by tissue-based single-cell RNA sequencing, Molecular cancer, 19(1): 1-17.
https://doi.org/10.1186/s12943-020-01264-9
PMid:33032611 PMCid:PMC7545561
Mcgranahan N., and Swanton C., 2017, Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future, Cell, 168: 613-628.
https://doi.org/10.1016/j.cell.2017.01.018
PMid:28187284
Peng J., Sun B.F., Chen C.Y., Zhou J.Y., Chen Y.S., Chen H., and Wu W., 2019, Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma, Cell research, 29(9): 725-738.
https://doi.org/10.1038/s41422-019-0195-y
PMid:31273297 PMCid:PMC6796938
Papalexi E., and Satija R., 2018, Single-cell RNA sequencing to explore immune cell heterogeneity, Nature Reviews Immunology, 18(1): 35-45.
https://doi.org/10.1038/nri.2017.76
PMid:28787399
Picelli S., 2017, Single-cell RNA-sequencing: The future of genome biology is now, RNA Biol, 14(5): 637-650.
https://doi.org/10.1080/15476286.2016.1201618
PMid:27442339 PMCid:PMC5449089
Stanta G., and Bonin S., 2018, Overview on clinical relevance of intra-tumor heterogeneity, Frontiers in medicine, 5: 85.
https://doi.org/10.3389/fmed.2018.00085
PMid:29682505 PMCid:PMC5897590
Vallejos C., Risso D., Scialdone A., Dudoit S., and Marioni J., 2017, Normalizing single-cell RNA sequencing data: challenges and opportunities, Nature Methods, 14: 565-571.
https://doi.org/10.1038/nmeth.4292
PMid:28504683 PMCid:PMC5549838
Wang Q., Wang Z., Zhang Z., Li C., Zhang M.M., Ye Y.J., Wang S., and Jiang K.W., 2020, Overview of the Technology of Single Cell Sequencing, Chinese Journal of Medicinal Guide, 46(9): 2030-2036.
Wang R., Zhao W., Yang H., Dong J., Lin W., He F., Cui M., and Zhou Z., 2021, Single-Cell RNA Sequencing Analysis of the Heterogeneity in Gene Regulatory Networks in Colorectal Cancer, Frontiers in Cell and Developmental Biology, 9: 765578.
https://doi.org/10.3389/fcell.2021.765578
PMid:34917613 PMCid:PMC8669944
Welch D., 2016, Tumor Heterogeneity--A' Contemporary Concept' Founded on Historical Insights and Predictions, Cancer Research, 76(1): 4-6.
https://doi.org/10.1158/0008-5472.CAN-15-3024
PMid:26729788 PMCid:PMC4773023
Yang S., Wang M., Hua Y., Li J., Zheng H., Cui M., Huang N., Liu Q., and Liao Q., 2024, Advanced insights on tumor-associated macrophages revealed by single-cell RNA sequencing: The intratumor heterogeneity, functional phenotypes, and cellular interactions, Cancer Lett, 584: 216610..
https://doi.org/10.1016/j.canlet.2024.216610
PMid:38244910
Ye X., Zhang W., Futamura Y., and Sakurai T., 2020, Detecting Interactive Gene Groups for Single-Cell RNA-Seq Data Based on Co-Expression Network Analysis and Subgraph Learning, Cells, 9(9): 1938.
https://doi.org/10.3390/cells9091938
PMid:32825786 PMCid:PMC7563496
Zhang X.C., and Wang X.P., 2019, Progress of single-cell sequencing in solid tumors, China Oncology, 29(7): 535-539.
https://doi.org/10.1360/TB-2019-0202
Zhang Y., Wang D., Peng M., Tang L., Ouyang J., Xiong F., and Xiong W., 2021, Single‐cell RNA sequencing in cancer research, Journal of Experimental & Clinical Cancer Research, 40: 1-17.
https://doi.org/10.1186/s13046-021-01874-1
PMid:33648534 PMCid:PMC7919320
Zeisel A., Hochgerner H., and Lonnerberg P., 2018, Molecular architecture of the mouse nervous system, Cell, 174(4): 999-1014.
https://doi.org/10.1016/j.cell.2018.06.021
PMid:30096314 PMCid:PMC6086934
Zheng L., Qin S., Si W., Wang A., Xing B., Gao R., Ren X., Wang L., Wu X., Zhang J., Wu N., Zhang N., Zheng H., Ouyang H., Chen K., Bu Z., Hu X., Ji J., and Zhang Z., 2021, Pan-cancer single-cell landscape of tumor-infiltrating T cells, Science, 374(6574): abe6474.
https://doi.org/10.1126/science.abe6474
PMid:34914499
. PDF(407KB)
. HTML
Associated material
. Readers' comments
Other articles by authors
. Tao Chen
Related articles
. Tumor heterogeneity
. Single-cell RNA Sequencing
. Cell Subpopulation
. Gene Expression
. Tumor Cell Interactions
Tools
. Email to a friend
. Post a comment