Review and Progress
Mechanisms of Interaction Between Epigenetic Modifications and Cancer Genomics 
 Author
Author  Correspondence author
Correspondence author
Cancer Genetics and Epigenetics, 2024, Vol. 12, No. 2
Received: 08 Mar., 2024 Accepted: 15 Apr., 2024 Published: 27 Apr., 2024
The interaction mechanism between epigenetic modifications and cancer genomics is a highly anticipated topic in the current field of cancer research. With the continuous advancement of technology, researchers' in-depth understanding of these two fields provides new perspectives for revealing the occurrence and development of cancer. Epigenetic modifications and cancer genomics play a crucial regulatory role in cell fate, gene expression, and tumorigenesis. The application of high-throughput sequencing technology enables researchers to have a more comprehensive understanding of the distribution and impact of epigenetic modifications in the cancer genome. However, the current challenges faced by cancer research, such as cancer heterogeneity and treatment resistance, further indicate the urgency of in-depth research on this interaction mechanism. This review introduces the basic concepts of cancer genomics and epigenetic modifications, explores the regulatory role of epigenetic modifications in the occurrence and development of cancer, and discusses the mutual influence between epigenetic modifications and cancer genomics. The aim is to deepen the understanding of the molecular mechanisms of cancer and provide scientific basis for precision medicine and personalized treatment.
1 Introduction
In the field of contemporary medical research, cancer remains a highly focused topic. As a complex and diverse disease, the mechanisms of cancer development have always been a focal point for scientists. With the advancement of high-throughput sequencing technologies, research in cancer genomics has gradually unveiled the genomic abnormalities in cancer cells, such as gene mutations and chromosomal rearrangements. This field's development provides a theoretical foundation for personalized cancer treatment. However, relying solely on genomics cannot fully explain the complexity of cancer.
Epigenetic modifications, as a key mechanism in gene expression regulation, have garnered significant interest from researchers. Epigenetic modifications include various forms such as DNA methylation, histone modifications, and non-coding RNA, which can rapidly and dynamically adjust gene expression levels (Wang et al., 2021). Therefore, understanding the regulatory role of epigenetics in cancer and its interaction with genomics is crucial for revealing the comprehensive mechanisms of cancer.
Research indicates that cancer development is a multifactorial and multistep process (Zou et al., 2019), involving the combined effects of gene mutations, epigenetic modifications, environmental factors, and more. Amidst this complexity, the interplay between cancer genomics and epigenetics has become a highly anticipated research direction. Cancer cells often exhibit genomic instability, and abnormal epigenetic modifications may be one of the critical reasons for this instability.
Although significant progress has been made in the independent studies of cancer genomics and epigenetics, understanding how they interact and their roles throughout cancer development remains limited. In-depth research into these interaction mechanisms will aid in a more comprehensive understanding of cancer complexity and provide new insights for future therapeutic strategies. By reviewing existing literature and studies, this research aims to elucidate the close relationship between these two levels and explore their significance in cancer occurrence, development, and treatment. This study aims to provide references for further exploration in the field of cancer research, offering a comprehensive perspective to researchers and clinicians and promoting the advancement of personalized and precise cancer treatment.
2 Overview of Cancer Genomics
2.1 Characteristics of cancer genomes
Research in cancer genomics has revealed a series of characteristics at the genomic level of tumor cells, highlighting genetic variations and genomic instability in the development of cancer. These features collectively depict a highly complex, heterogeneous, and evolving genomic landscape. Cancer cells often accumulate a significant number of gene mutations, including point mutations, deletions, insertions, and copy number variations. These mutations can affect key regulatory genes and signaling pathways, driving cellular transformation. The chromosomal structure of cancer cells frequently undergoes major variations, such as chromosomal breaks, rearrangements, and deletions. These structural variations lead to gene misplacement and recombination, further promoting tumor formation.
Cancer cells often exhibit genomic instability, manifesting as abnormalities in chromosome number and structure during cell division. This instability contributes to the diversity and evolution of tumor cells. Cancer genomics research has found that the activation of oncogenes and the inactivation of tumor suppressor genes are major drivers of cancer development (Kontomanolis et al., 2020). Mutations in these genes directly affect critical processes such as cell growth, differentiation, and apoptosis. Genomic variations are influenced not only by internal cellular factors but also by the tumor microenvironment. This includes interactions between tumor cells and surrounding tissues, the immune system, and blood vessels, further shaping the genomic characteristics of tumors.
2.2 Classification and function of cancer-related genes
Cancer genomics research has discovered that cancer development is closely related to the abnormal expression and mutations of genes involved in regulating key processes such as cell growth, apoptosis, repair, and differentiation. These cancer-related genes can be broadly classified into two main categories: oncogenes and tumor suppressor genes.
Oncogenes include genes that activate cell proliferation, maintain genomic stability, and inhibit apoptosis. Genes that activate cell proliferation include various growth factor receptors and signaling molecules such as the EGFR (epidermal growth factor receptor) and the RAS (Rat Sarcoma viral oncogene homolog) family. Mutations or overexpression of these genes can lead to uncontrolled cell proliferation. Genes that maintain genomic stability include DNA repair-related genes like BRCA1 and BRCA2 (Huang et al., 2022). Mutations in these genes can impair DNA repair mechanisms, increasing the risk of cancer. Genes that inhibit apoptosis, such as members of the BCL2 family, promote cell survival by inhibiting the apoptotic process. Their overexpression can lead to cells evading programmed death and developing into cancer cells.
Tumor suppressor genes include genes that regulate the cell cycle, participate in apoptosis, and maintain genomic stability. Genes that regulate the cell cycle, such as TP53 (tumor protein 53) and p16INK4a, prevent abnormal cell proliferation by controlling cell cycle progression. Genes involved in apoptosis, such as APC (adenomatous polyposis coli) and PTEN, guide cells towards programmed death when abnormalities occur. Genes that maintain genomic stability, such as ATM (ataxia-telangiectasia mutated) and CHK2 (checkpoint kinase 2), prevent genomic instability by monitoring and repairing DNA damage. These genes play crucial regulatory roles in normal cells, but when they mutate or are abnormally expressed, they can cause cells to lose their normal growth and regulatory mechanisms, thereby promoting cancer.
2.3 Development and application of genomic technologies
In recent years, with the rapid development of genomic technologies, researchers have made significant progress in understanding cancer genomes. The breakthrough of next-generation sequencing (NGS) technology is crucial for obtaining comprehensive genomic information from cancer patients. Technologies such as whole-exome sequencing (WES) and whole-genome sequencing (WGS) have enabled researchers to reveal the genetic variations in cancer more comprehensively.
Epigenetic technologies have also made significant progress. Techniques such as methylation arrays and whole-genome bisulfite sequencing (WGBS) efficiently detect DNA methylation levels, uncovering epigenetic variations in cancer. ChIP-Seq (chromatin immunoprecipitation sequencing) technology analyzes the interaction between proteins and DNA, providing researchers with an important tool to study the regulatory mechanisms of histone modifications in cancer.
The rise of single-cell sequencing technology allows for a more detailed understanding of tumor heterogeneity and genomic variations at the single-cell level. Single-cell RNA sequencing (scRNA-seq) reveals potential differences among tumor cell populations, while single-cell DNA sequencing (scDNA-seq) uncovers genomic variations at the individual cell level. The application of big data analysis and artificial intelligence enables genomics research to handle large datasets more effectively. The integration of these technologies not only enhances researchers' deep understanding of cancer genomics but also provides strong support for personalized medicine and precision therapy.
3 Basic Concepts of Epigenetic Modifications
3.1 DNA methylation
DNA methylation is a key epigenetic modification that regulates gene expression by adding methyl groups to the DNA molecule (Pan et al., 2021). This modification occurs on the cytosine rings of DNA molecules, specifically at the C5 position of cytosine nucleotides. The methylation process primarily involves the DNA methyltransferases (DNMTs) family and demethylases (Figure 1). DNMTs are responsible for transferring methyl groups onto DNA, while demethylases are capable of removing methyl groups from DNA.
|
Figure 1 Mechanism of DNA Methylation |
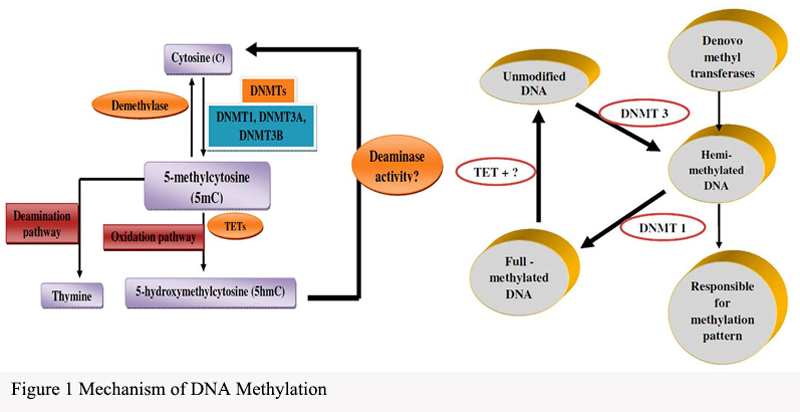
This modification pattern exhibits significant dynamism, varying not only among different cell types and stages of life but also demonstrating the ability to adjust dynamically in disease states. Particularly in CpG islands (regions rich in CpG dinucleotides), DNA methylation patterns are especially prominent. These islands are usually located in gene promoter regions, directly influencing the transcriptional activity of adjacent genes.
DNA methylation affects gene expression through various mechanisms, including blocking the binding of transcription factors and recruiting methylation recognition proteins to influence chromatin structure. This impact mechanism can be both stable and reversible, providing multiple levels of regulation for gene expression. In cancer, abnormal changes in DNA methylation are a common phenomenon. Cancer cells typically exhibit a genome-wide hypomethylation state, especially in CpG island regions. This hypomethylation state may lead to the inactivation of certain tumor suppressor genes and the overactivation of oncogenes, thereby driving tumor development.
3.2 Histone modifications
Histone modification is another important form of epigenetic modification that regulates gene accessibility and transcriptional activity by altering the structure and compactness of chromatin. This modification involves various covalent modifications on histones, including methylation, acetylation, and phosphorylation (Figure 2). Lysine residues on histones can be methylated, particularly on the side chains of lysine. This modification is often associated with gene silencing, as the accumulation of methylation at specific histone sites can lead to a condensed and inactive state of the gene locus.
|
Figure 2 Factors of Histone Modifications |
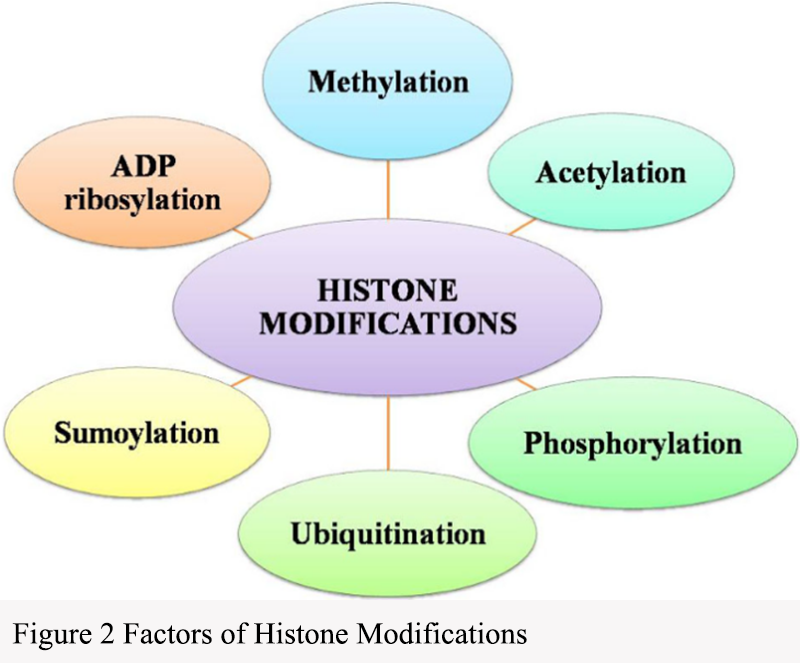
Lysine residues on histones can also be acetylated, which weakens the interaction between histones and DNA by removing positive charges. This modification is usually associated with gene activation, promoting chromatin relaxation and gene transcription. Histone phosphorylation can regulate chromatin structure and affect its compactness. Phosphorylation events are often closely related to cell signaling and cell cycle regulation. Ubiquitination, the attachment of small ubiquitin proteins to histones, can also alter chromatin structure and influence gene expression. These histone modifications form a complex network within the cell nucleus, regulating gene expression and cellular function.
3.3 The role of Non-coding RNA
Non-coding RNA (ncRNA) are RNA molecules that do not encode proteins but play critical roles in gene expression and cellular function. They are widely involved in the regulation of epigenetic modifications, impacting gene regulation, maintenance of chromatin structure, and various cellular biological processes.
miRNAs are short non-coding RNAs, approximately 20-22 nucleotides long, that regulate gene translation and degradation by binding to the mRNA of target genes. Abnormal expression of miRNAs is closely related to the occurrence and development of various cancers, and they can act as regulators of tumor suppressor genes or oncogenes.
siRNA is a type of non-coding RNA that regulates gene expression through RNA interference (RNAi). siRNA can silence target genes by specifically binding to and mediating the degradation of target mRNA. In cancer research, siRNA technology is widely used for gene silencing and the development of therapeutic strategies.
lncRNAs are long non-coding RNAs that can exceed 200 nucleotides in length. lncRNAs regulate gene expression through various mechanisms, including chromatin remodeling, transcriptional regulation, and competitive binding with miRNAs. In cancer, abnormal expression of many lncRNAs is closely associated with tumor development and progression.
circRNAs are closed circular RNA molecules with resistance to degradation. They can act as "sponges" for miRNAs, sequestering miRNAs and preventing them from binding to target gene mRNAs, thus influencing gene expression. These ncRNAs participate in multiple aspects of cancer, including cell cycle regulation, proliferation, and apoptosis, through complex interaction networks.
3.4 Other epigenetic modification mechanisms
In addition to DNA methylation, histone modifications, and non-coding RNA, there are other epigenetic modification mechanisms that together form a complex network regulating gene expression and cellular functions. Glycosylation is a modification process involving the attachment of sugar molecules to proteins or nucleic acids (Yang et al., 2021). Glycosylation modifications are involved in key processes in cancer, such as cell adhesion, signal transduction, and the cell cycle, influencing the proliferation and invasion capabilities of tumor cells.
Beyond phosphorylation on histones, many other proteins in cells can be regulated through phosphorylation. Phosphorylation modifications participate in the regulation of various signaling pathways, impacting key processes such as cell survival, proliferation, and apoptosis. Protein acetylation, besides histone acetylation, is also a common epigenetic regulatory mechanism. Acetylation modifications regulate protein stability and function, influencing biological processes such as metabolism, cell cycle, and apoptosis.
Ubiquitination modifies other proteins by attaching small ubiquitin proteins. This modification process is involved in crucial processes such as protein degradation, signal transduction, and DNA repair, playing an essential role in maintaining cellular homeostasis and function. Enzymatic modifications, such as those by kinases and phosphatases, regulate protein phosphorylation states and participate in the regulation of multiple cellular signaling pathways. These epigenetic modification mechanisms collectively construct a complex and dense regulatory network, enabling cells to respond to internal and external environmental changes and adjust gene expression and biological processes. In cancer, abnormal changes in these modifications can lead to gene dysregulation, thereby promoting tumor development.
4 The Role of Epigenetics in Cancer Development and Progression
4.1 Abnormal DNA methylation and its relationship with tumors
Abnormal DNA methylation is one of the common molecular events in cancer development and progression. This abnormality typically manifests as global hypomethylation and hypermethylation in certain CpG island regions, significantly impacting gene expression and genomic stability. In cancer cells, global hypomethylation is a widespread phenomenon. This hypomethylation state can lead to genomic instability, promoting abnormal variations in key genomic regions, including gene rearrangements and mutations. This instability may be one of the driving forces of cancer development.
CpG islands are gene promoter regions rich in CpG dinucleotides. Under normal circumstances, these regions are usually methylated. However, in cancer, abnormal hypermethylation in certain CpG island regions can lead to the silencing of associated genes. These genes are often critical in inhibiting cell proliferation, regulating apoptosis, or participating in DNA repair, and their inactivation may promote cancer development (Casalino and Verde, 2020).
DNA repair is a crucial process for maintaining genomic stability, and abnormal methylation of DNA repair genes is closely related to cancer. For example, BRCA1 and BRCA2 are genes involved in DNA repair, and their abnormal methylation can impair DNA repair functions, increasing the risk of hereditary breast and ovarian cancers. Abnormal DNA methylation not only plays a key role in cancer development but may also serve as a biomarker for early cancer events. Measuring DNA methylation levels in blood or tissue can provide clues for early cancer diagnosis and treatment.
4.2 Changes in histone modifications and their association with cancer progression
Abnormal changes in histone modifications play a critical role in cancer progression, affecting gene expression, cell cycle regulation, and chromatin structure, thereby driving cancer development and metastasis (Ilango et al., 2020). Histone methylation is an important form of histone modification that influences gene accessibility and transcriptional activity by altering chromatin structure and stability (Figure 3). In cancer, hypermethylation at certain gene loci can lead to the inactivation of tumor suppressor genes, while the hyperactivation of certain oncogenes is also closely associated with abnormal changes in methylation.
|
Figure 3 Histone Methylation and Gene Expression |
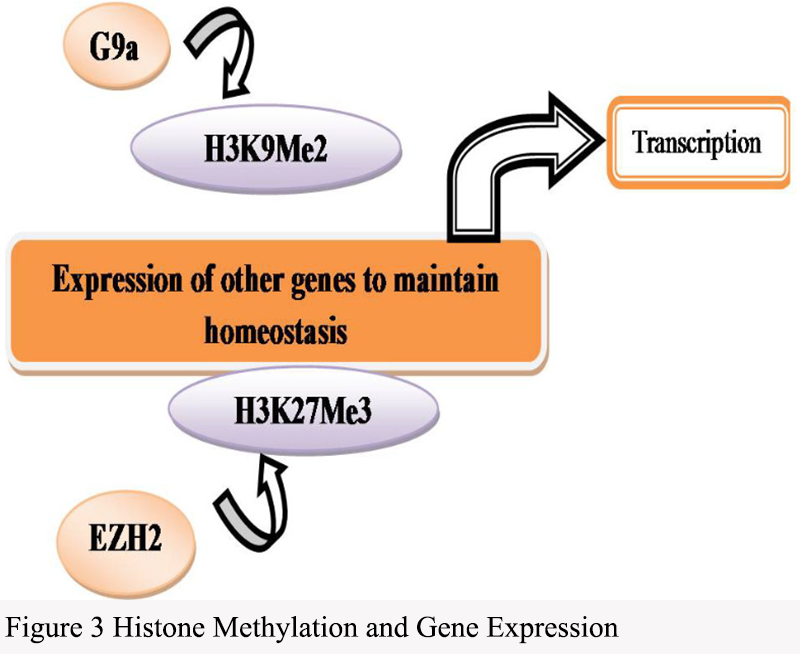
.png){kind=link}
.png){kind=link}
.png){kind=link}
Histone acetylation is usually associated with gene activation. In cancer, abnormal acetylation of certain genes may lead to their overactivation, promoting cancer cell proliferation and survival. Conversely, dysregulation of histone acetylation can also lead to the inactivation of tumor suppressor genes. Histone phosphorylation is involved in multiple key biological processes such as cell signaling, cell cycle regulation, and gene expression. In cancer, abnormal histone phosphorylation can disrupt cellular signals, increasing the proliferation and metastatic potential of cancer cells.
Histone ubiquitination is a modification process that regulates histones by attaching small ubiquitin proteins. In cancer, abnormal ubiquitination can lead to the degradation of certain tumor suppressor proteins, thereby promoting uncontrolled cancer cell growth. Some histone proteolytic modifications, such as those by protein kinases and proteases, are involved in regulating multiple cell signaling pathways. In cancer, abnormal changes in these enzymatic modifications can cause cell cycle dysregulation and evasion of apoptosis.
4.3 Regulatory mechanisms of Non-coding RNA in tumorigenesis
Non-coding RNAs (ncRNAs) play key regulatory roles in the development and progression of tumors, including miRNA, lncRNA, siRNA, and circRNA. These ncRNAs participate in multiple biological processes of tumors through various mechanisms. miRNAs, as short non-coding RNAs, regulate gene expression by binding to the mRNA of target genes. In tumors, abnormal expression of certain miRNAs leads to aberrant expression of target genes, thereby promoting or inhibiting tumor growth and metastasis. miRNAs also regulate multiple cell signaling pathways, affecting cell proliferation, apoptosis, and invasion.
lncRNAs regulate chromatin structure and stability by interacting with chromatin, thus influencing gene accessibility. In tumors, abnormal expression of certain lncRNAs may lead to the silencing of tumor suppressor genes or the overexpression of oncogenes. Additionally, some lncRNAs act as "sponges" for miRNAs, binding to miRNAs and mitigating their negative regulation on target genes.
siRNA, as part of the RNA interference (RNAi) pathway, achieves gene silencing by specifically degrading the mRNA of target genes. In cancer therapy, siRNA technology is widely used to inhibit the expression of oncogenes. circRNAs act as "sponges" for miRNAs, absorbing miRNAs and preventing them from negatively regulating target genes, thereby affecting cellular biological processes. Moreover, some circRNAs interact with proteins, regulating protein function.
5 Interrelationship Between Cancer Genomics and Epigenetics
Cancer genomics and epigenetics are two critical aspects of understanding cancer complexity, with a close interrelationship between them. Cancer genomics focuses on genomic variations and mutations, while epigenetics studies changes in gene expression and regulatory levels. This interrelationship plays a vital role in revealing cancer pathogenesis, discovering potential therapeutic targets, and developing personalized treatment plans.
5.1 Current status of research on the relationship between cancer genomics and epigenetics
Research on the relationship between cancer genomics and epigenetics occupies an important position in current cancer research, providing extensive information for understanding cancer pathogenesis, identifying potential therapeutic targets, and developing personalized treatment strategies. In recent years, researchers have been integrating multi-omics data from cancer genomics and epigenetics, including gene mutations, gene expression, DNA methylation, and histone modifications (Ilango et al., 2020). This comprehensive analysis helps to fully understand the biological characteristics of tumors and reveal the interactions between genomics and epigenetics.
Researchers have successfully identified subtypes of various cancers through integrated analysis of genomic and epigenetic data. This classification aids in a better understanding of cancer heterogeneity and guides the development of personalized treatment plans. Correlation studies have revealed new therapeutic targets that may involve specific gene mutations and epigenetic regulatory pathways. Researchers are exploring therapeutic strategies targeting these new targets, providing new directions for innovative drug development.
By tracking the genomic evolution and dynamic epigenetic changes of tumors, researchers can gain a deep understanding of the tumor evolution process. This is important for understanding the formation of drug resistance, metastasis, and therapeutic resistance mechanisms. By combining cancer genomics and epigenetic information, researchers are gradually achieving the formulation of personalized treatment strategies. Based on the unique molecular characteristics of patients, more precise treatment plans can be chosen to improve treatment efficacy.
5.2 Epigenetic modifications of cancer-related genes
Cancer development and progression are often accompanied by abnormal gene expression, and epigenetic modifications play a key role in this process as an important gene regulatory mechanism. For example, TP53, a tumor suppressor gene (Yuan et al., 2020), often experiences abnormal DNA methylation in its promoter region in various cancers, leading to gene silencing or repressed expression. Additionally, abnormal changes in histone acetylation and methylation can also affect the chromatin structure of the TP53 gene, thus influencing its function in cancer.
BRCA1 and BRCA2 are genes associated with breast and ovarian cancers, and abnormal DNA methylation in their promoter regions is closely related to the occurrence of these cancers, potentially leading to gene silencing. Histone acetylation and methylation also play key roles in the regulation of these two genes, with abnormal modifications potentially causing changes in gene expression. RB1, a gene associated with retinoblastoma, commonly experiences promoter region DNA methylation associated with various cancers, leading to downregulation of gene expression. Changes in histone methylation and acetylation may also play a role in the inactivation of the RB1 gene.
PTEN, a tumor suppressor gene, has been shown to have DNA methylation associated with various cancers, potentially leading to gene silencing or downregulation of expression. Abnormal changes in histone acetylation and phosphorylation can affect the expression and function of the PTEN gene. EGFR, an epidermal growth factor receptor, has DNA methylation associated with various cancers, potentially leading to gene silencing or downregulation of expression. Histone acetylation and phosphorylation may play roles in the regulation of the EGFR gene, affecting the proliferation and survival of cancer cells. These examples reveal the diverse epigenetic modifications of cancer-related genes. In-depth research on these modifications is crucial for understanding cancer's molecular mechanisms, discovering new therapeutic targets, and developing personalized treatment strategies.
5.3 Challenges and opportunities in epigenetics in cancer genomics research
Epigenetics plays a critical role in cancer research, yet its study faces a series of challenges. Epigenetics research involves multidimensional data such as genomics, transcriptomics, and methylomics, requiring interdisciplinary collaboration and advanced analytical tools to handle the complexity of data integration. Additionally, the rapid development of technological platforms increases the burden on researchers to learn and adapt to new methods. A deep understanding of biological complexity is a prerequisite for interpreting epigenetic data, necessitating profound biological knowledge. The heterogeneity of clinical samples is a common issue that can affect the reliability of research results.
Despite these challenges, epigenetics research offers vast opportunities for cancer treatment. In-depth research in epigenetics provides opportunities for the development of precision medicine. Epigenetic analysis of patient samples allows for a better understanding of tumor molecular characteristics, providing a basis for personalized treatment strategies. Epigenetics research is expected to reveal new therapeutic targets, offering new directions for developing more effective anticancer drugs. With the continuous advancement of bioinformatics technology, methods for data integration and analysis are also improving. Integrating epigenetic and genomic data will provide support for a more comprehensive understanding of cancer's molecular mechanisms. The application of new technologies, such as single-cell sequencing and spatial transcriptomics, offers more detailed and comprehensive tools for epigenetic research, potentially addressing some of the technical challenges.
6 Conclusion
In the process of cancer development and progression, there is a complex and close interplay between genomics and epigenetics. Mutations and variations in genomics not only directly affect gene sequences but also influence epigenetic modifications. For instance, abnormal changes in epigenetic modifications, such as DNA methylation and histone modifications of cancer-related genes, are often correlated with genomic variations (Zhang et al., 2022), collectively driving tumor development.
Epigenetics plays a crucial role in the mechanisms of cancer pathogenesis, influencing gene expression and regulation. Abnormal changes in modifications such as DNA methylation and histone modifications can lead to the inactivation of tumor suppressor genes or the overactivation of oncogenes, promoting uncontrolled proliferation of cancer cells. Additionally, non-coding RNAs, such as miRNAs and lncRNAs, are also involved in the interplay between epigenetics and genomics. They regulate gene transcription and translation levels, affecting the functions and biological behaviors of tumor cells.
Personalized treatment strategies targeting specific epigenetic modifications and genomic variations will become a major trend in cancer therapy. By gaining an in-depth understanding of the epigenetic and genomic characteristics of each patient’s tumor, doctors can formulate more precise treatment plans for patients. Further research will help discover more novel therapeutic targets associated with the interplay between epigenetics and genomics. The discovery of these targets will provide new directions for the development of new drugs and therapeutic strategies.
With continuous technological advancements, integrative analysis of multi-omics data will become more comprehensive, encompassing more layers of information, and providing deeper insights into the mechanisms of complex diseases. Research on the association between epigenetics and cancer genomics is expected to help identify more specific biomarkers, enhancing the accuracy of early cancer diagnosis and prognosis assessment. The interplay between epigenetics and cancer genomics will drive cancer research and treatment towards deeper and more precise stages in the future. This offers a promising prospect for achieving personalized medicine, precision therapy, and ultimately overcoming cancer.
Conflict of Interest Disclosure
The author affirms that this research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Casalino L., and Verde P., 2020, Multifaceted roles of DNA methylation in neoplastic transformation, from tumor suppressors to EMT and metastasis, Genes, 11(8): 922.
https://doi.org/10.3390/genes11080922
PMid:32806509 PMCid:PMC7463745
Huang F., Chen X.N., Yu L., Jiang H.Q., Shi T.Y., Shen M.N., Zhang C.Y., Pan B.S., Wang B.L., and Guo W., 2022, Clinical characteristics of germline BRCA 1/2 mutations in patients with breast cancer, Fudan Xuebao Yixueban (Fudan University Journal of Medical Sciences), 49(6): 891-897.
Ilango S., Paital B., Jayachandran P., Padma P.R., and Nirmaladevi R., 2020, Epigenetic alterations in cancer, Frontiers in Bioscience-Landmark, 25(6): 1058-1109.
PMid:32114424
Kontomanolis E.N., Koutras A., Syllaios A., Schizas D., Mastoraki A., Garmpis N., Diakosavvas M., Angelou K., Tsatsaris G., Pagkalos A., Ntounis T., and Fasoulakis Z., 2020, Role of oncogenes and tumor-suppressor genes in carcinogenesis: a review, Anticancer Research, 40(11): 6009-6015.
https://doi.org/10.21873/anticanres.14622
PMid:33109539
Pan H., Renaud L., Chaligne R., Bloehdorn J., Tausch E., Mertens D., Fink A.M.; Fischer K., Zhang C., Betel D., Gnirke A., Imielinski M., Moreaux J., Hallek M., Meissner A., Stilgenbauer S., Wu C.J., Elemento O., and Landau D.A., 2021, Discovery of candidate DNA methylation cancer driver genes, Cancer Discovery, 11(9): 2266-2281.
https://doi.org/10.1158/2159-8290.CD-20-1334
PMid:33972312 PMCid:PMC8419066
Wang S.S., Xu J., Ji K.Y., and Hwang C.I., 2021, Epigenetic alterations in pancreatic cancer metastasis, Biomolecules, 11(8): 1082.
https://doi.org/10.3390/biom11081082
PMid:34439749 PMCid:PMC8394313
Yang W.B., Xu Y., Zhuo S.X., Wang X.Y., Li Y.J., Guo Y.F., Zhang Z.G., and Guo Y.Y., 2021, Progress of long non-coding RNAs related epigenetic modifications in cancer, Zhongguo Shengwu Gongcheng Zazhi (China Biotechnology), 41(8): 59-66.
Yuan L., Wang L., Du X., Qin L., Yang M., Zhou K., Wu M.P., Yang Y., Zheng Z.Y., Xiang Y., Qu X.P., Liu H.J., Qin X.Q., and Liu C., 2020, The DNA methylation of FOXO3 and TP53 as a blood biomarker of late-onset asthma, Journal of Translational Medicine, 18(1): 1-13.
https://doi.org/10.1186/s12967-020-02643-y
PMid:33298101 PMCid:PMC7726856
Zhang M., Yang L.L., Jia Y.L., and Wang T.Y., 2022, Research progress in the roles of DNA and histone methylations in epigenetic regulation, Shengwu Jishu Tongbao (Biotechnology Bulletin), 38(7): 23-30.
Zou T.H., Liao X.H., and Chen Y.X., Early warning, early diagnosis and prevention of carcinogenesis of chronic atrophic gastritis, Linchuang Huicui (Clinical Focus), 34(5): 407.
(1).png)
. FPDF(win)
. FPDF(mac)
. HTML
. Online fPDF
Associated material
. Readers' comments
Other articles by authors
. Jie Wang
Related articles
. Epigenetic modifications
. Cancer genomics
. DNA methylation
. Cancer driving genes
. Individualized treatment
Tools
. Post a comment